Korean Medical Device Approval Process
The elements of manufacturing and distribution of medical devices can be simplified as follows: manufacturer, seller, and product.
To sell medical devices in Korea, approval for all three elements mentioned above must be obtained. The required types of approvals are as follows:
- Distributor: Manufacturing/Import License
- Manufacturer (Factory): KGMP
- Product: Product Registration The detailed requirements for each element are as follows:
The detailed requirements for each element are as follows:
1. Manufacturing/Importing License
- This license is granted to medical device manufacturers or importers. To apply for the license, the appointment of a quality manager is mandatory. The quality manager must meet the qualifications specified in Article 11 of the Enforcement Rules of the Medical Device Act. Qualifications for the Quality Manager (Korean), Qualifications of the quality manager (English)
- Additionally, the appropriate quality system must be maintained, as defined by the Medical Device Manufacturing and Quality Management Standards and the requirements for importers.
- For detailed information, refer to the relevant regulations: Compliance Requirements for Importers (Korean) Compliance Requirements for Importers (English)
- Review Authority: Local Food and Drug Administration (FDA) offices.
2. KGMP Certification
- This refers to the audit of the quality management system for medical device manufacturers and contract manufacturers, which may be either a local inspection or a document review.
- If the manufacturer holds MDSAP certification, the initial audit may be substituted with a document review.
- Review Authority: Korea Food and Drug Administration (MFDS) and designated FDA-recognized inspection agencies (e.g., KTL, KTR, KCL, KTC).
3. Product Approval
- Depending on the medical device's classification, this process is called product registration, product certification, or product approval. The safety and performance of the device to be manufactured or imported are evaluated.
- Documentation related to the safety and performance of the product (e.g., IEC 60601, ISO 10993, clinical trial reports) must be submitted.
- Review Authority: Korea Food and Drug Administration (MFDS) and designated FDA-recognized inspection agencies (e.g., KTL, KTR, KCL, KTC).
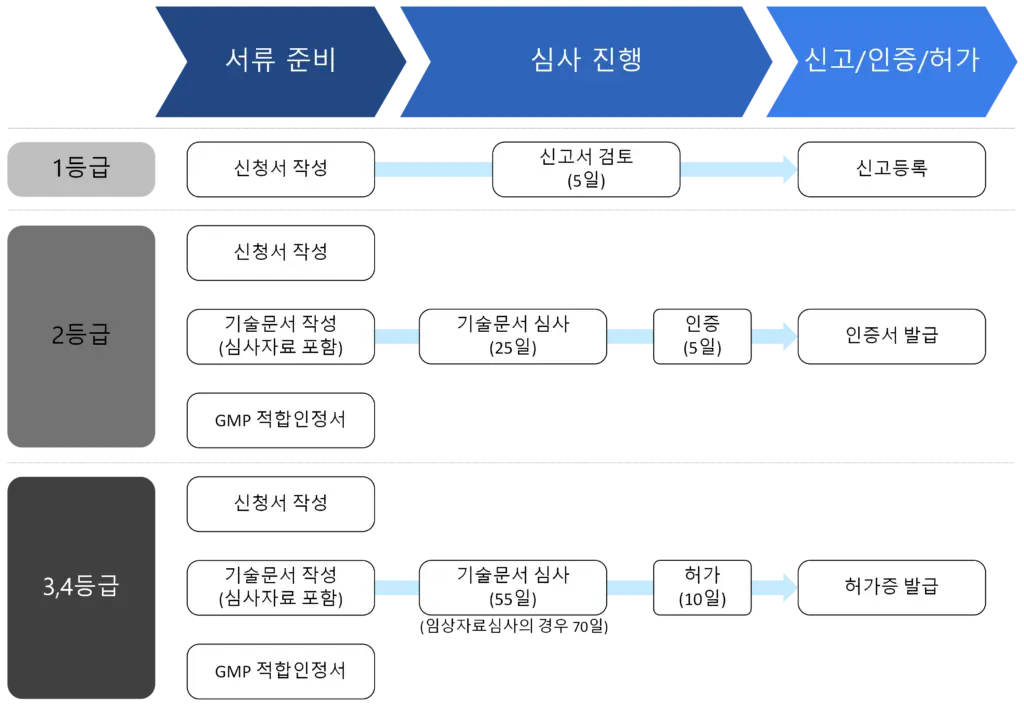
- The submission materials and processing time vary depending on the classification of the medical device being applied for:
- The validity period of the license is 5 years, and renewal of the license is required before the expiration date.