Focused on Quality, Driven by Results
MISSION-DRIVEN
의료기기의 제조 및 유통과정의 요소는 아래와 같이 제조자, 판매업자, 제품으로 단순화 할 수 있다.
한국에서 의료기기를 판매 하기 위해서는 상기 3가지 요소에 대한 허가를 모두 득해야 한다. 이 때, 필요한 허가의 종류는 다음과 같다:
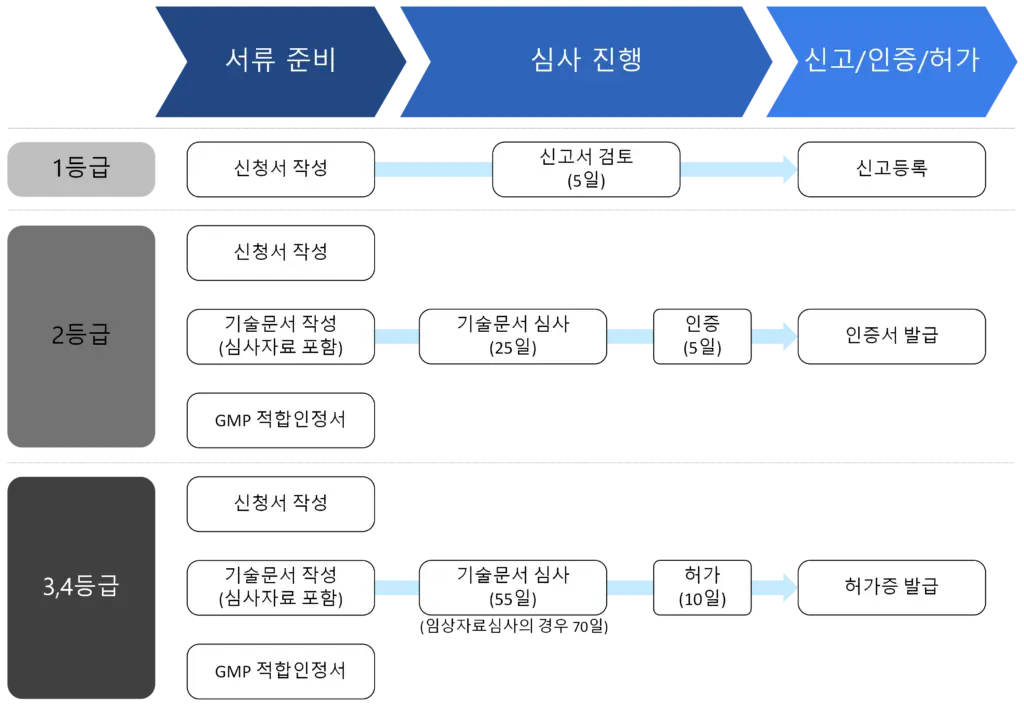
각 요소에 대한 세부 요구사항은 아래와 같다:
PATH TO SUCCCESS
전문가의 도움이 필요하신가요? 언제든지 문의해 주세요.