Focused on Quality, Driven by Results
MISSION-DRIVEN
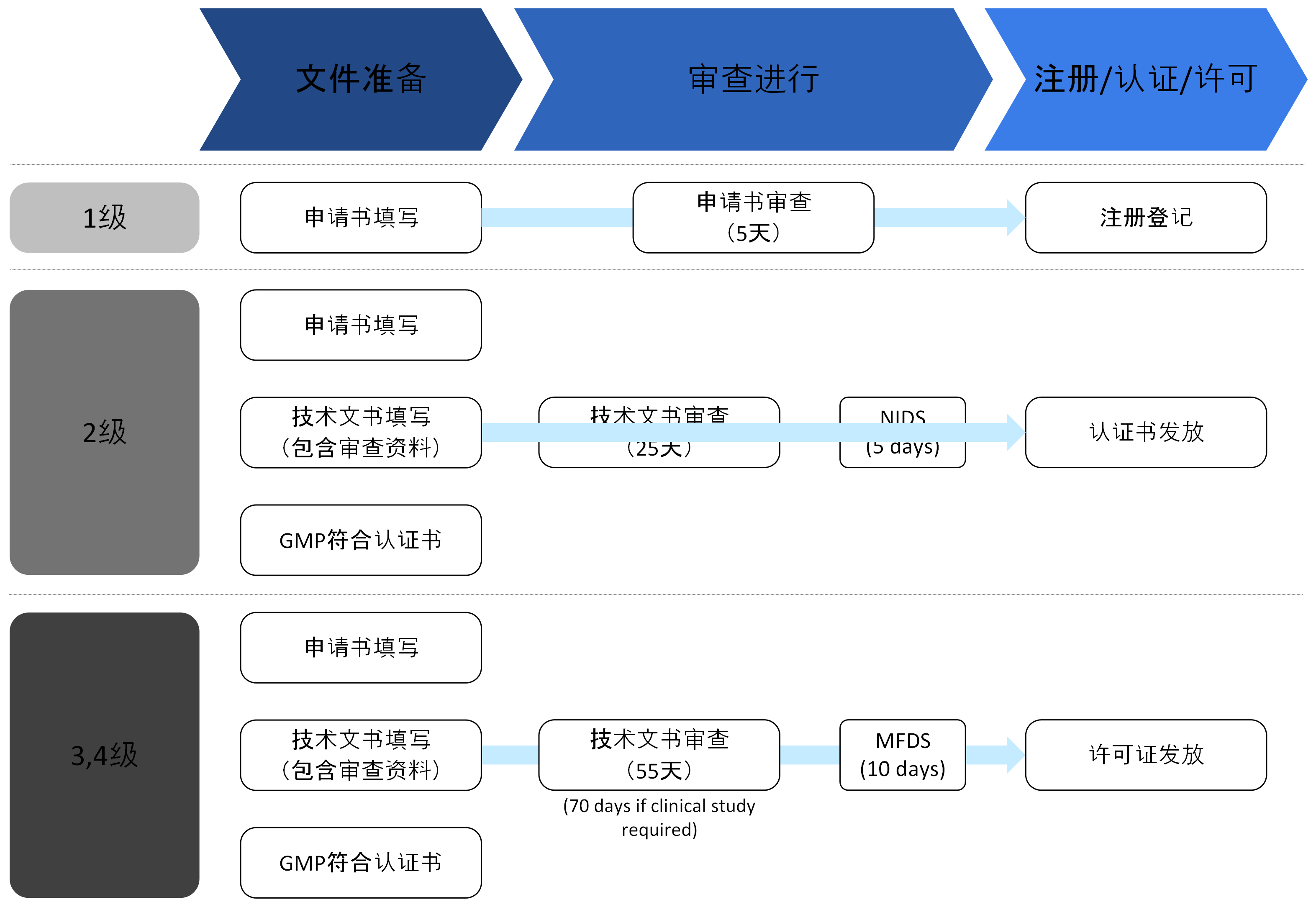
医疗器械的制造和流通过程的要素可以简化为以下三项:制造商、销售商和产品。
在韩国销售医疗器械,必须获得上述三项要素的批准。 所需的批准类型如下:
各个要素的详细要求如下:
PATH TO SUCCCESS
如果您需要专家的帮助,欢迎随时咨询。